

On May 6, 2024, the U.S. Food and Drug Administration published its anticipated final rule concerning oversight of laboratory-developed tests (LDTs). The final rule (i) affirms the FDA’s position that in vitro diagnostic products (IVDs) are devices under the Federal Food, Drug, and Cosmetic Act (FDCA), including when the manufacturer of the IVD is a laboratory; (ii) sets out FDA’s enforcement discretion phaseout policy for LDTs in stages; (iii) applies “targeted” enforcement discretion for certain categories of LDTs; and (iv) reiterates that certain other categories of LDTs, which receive either full enforcement discretion or no enforcement discretion at all, are not subject to the phaseout policy. Concurrently with the publication of the final rule, FDA also issued two draft guidance documents: (i) Consideration of Enforcement Policies for Tests During a Section 564 Declared Emergency, and (ii) Enforcement Policy for Certain In Vitro Diagnostic Devices for Immediate Public Health Response in the Absence of a Declaration under Section 564.
FDA defines LDTs as a subset of IVDs designed, manufactured, and used within a single laboratory certified under the Clinical Laboratory Improvement Amendments (CLIA) to perform high-complexity testing. FDA has stated that increased oversight of LDTs is necessary to protect patients and ensure the accuracy of diagnostic tests. According to FDA, many modern LDTs rely on increasingly high-tech instrumentation or software and are frequently used to guide critical decisions, yet scientific data collected over the years indicates that IVDs offered as LDTs do not always produce accurate or reliable results. Therefore, FDA believes that LDTs should be required to meet FDA requirements for medical devices to ensure patient safety.
Enforcement Discretion Phase-Out Policy for LDTs
Under the final rule, FDA will gradually phase out its exercise of enforcement discretion for LDTs in five stages over the next four years. Each stage contains a deadline for achieving certain regulatory requirements, such that by the Stage 5 deadline, all LDTs subject to oversight must be in full compliance with medical device requirements under the FDCA. All LDTs must comply with Medical Device Reporting requirements, reports of correction and removals, registration and listing requirements, and labeling requirements as part of the phaseout policy unless expressly excluded.
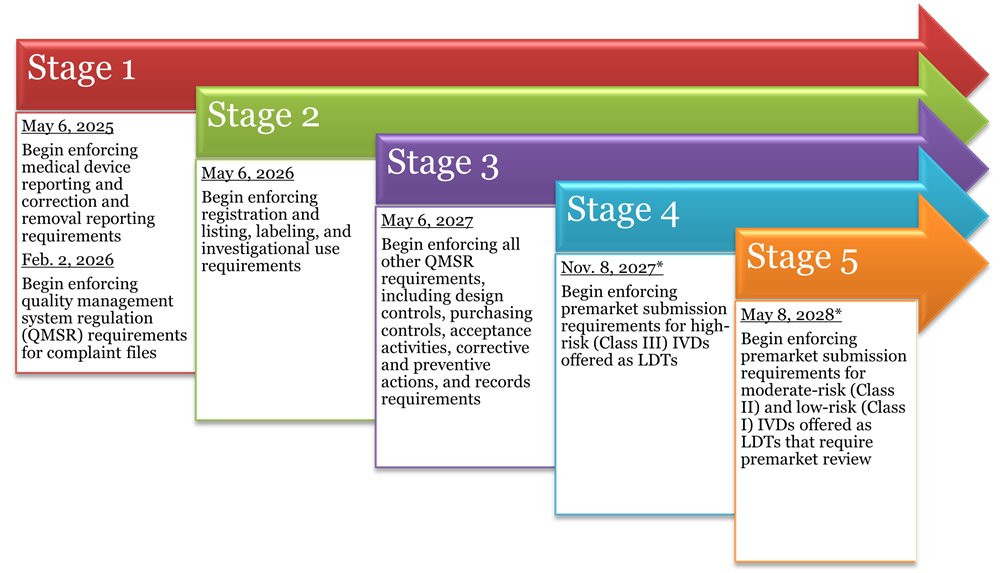
*As long as a premarket submission has been received by this date, FDA states it will continue to exercise enforcement discretion for the LDT for the pendency of its review.
Targeted Enforcement Discretion
Notably, the final rule applies “targeted” enforcement discretion to the following categories of LDTs as part of the phaseout policy. According to FDA, these limited exceptions were added to the final LDT rule in response to public comments on the proposed LDT rule.
- LDTs currently on the market that were first marketed prior to the issuance of the final rule on May 6, 2024, are not required to undergo premarket review or comply with most QMSR requirements (except with respect to records) if no significant modifications are made to the indications for use, operating principles, technologies, performance specifications, or safety specifications. All other phaseout policies apply. If the LDTs are modified (e.g., indications for use or operating principle modifications; modifications including a significantly different technology; or modifications that adversely impact the performance or safety specifications), they will be required to comply with premarket review and QS requirements.
- LDTs approved by the NY Department of Health’s Clinical Laboratory Evaluation Program (CLEP) are not required to undergo premarket review, if they are designed, manufactured, and used within a single CLIA-certified, high-complexity laboratory. This category includes LDTs introduced to the market subsequent to the issuance of the final rule (May 6, 2024) that are high-risk LDTs that receive full technical review and approval by CLEP, as well as moderate-risk LDTs that receive conditional approval if the approval is not subsequently withdrawn by CLEP upon a full technical review. All other phaseout policies apply.
- 510(k)-cleared or de novo-authorized LDTs modified by a CLIA-certified, high-complexity laboratory are not required to undergo premarket review, even if the 510(k) clearance or de novo authorization is held by a different manufacturer, if the modification is of a type that would not require the manufacturer to submit a new 510(k). All other phaseout policies apply.
- LDTs addressing an “unmet need” for patients of a health care system within which the laboratory is integrated are not required to undergo premarket review or comply with most QMSR requirements (except with respect to records) as long as either there is no FDA-authorized IVD for the particular disease or condition addressed by this LDT or there is such an IVD, but that IVD is not accessible to the patient or is not indicated for use on the patient. FDA intends to provide additional guidance to clarify remaining uncertainty about the definition of the term “unmet need.” Notably, this carveout does not apply to tests used in an affiliated hospital under different ownership from the hospital that developed the test. All other phaseout policies apply.
- Non-molecular antisera LDTs for rare red blood cell antigens are not required to undergo premarket review or comply with most QMSR requirements (except with respect to records) if the LDT is manufactured and performed by blood establishments (e.g., transfusion services and immunohematology laboratories) and when no alternative IVD is available to meet the patient’s need for a blood transfusion. All other phaseout policies apply.
.
LDTs Excluded from Phaseout Policy
The FDA intends to exercise full enforcement discretion for certain categories of LDTs, which continue to be exempt from all premarket review and quality requirements for medical devices under the FDCA or facility registration, as long as the LDTs are designed, manufactured, and used within a CLIA-certified, high-complexity laboratory:
- 1976-Type LDTs (i.e., LDTs that use only manual techniques and components legally marketed for clinical use);
- Human leukocyte antigen tests used in connection with organ, stem cell, and tissue transplantation;
- Tests manufactured and used within the U.S. Department of Defense or Veterans Health Administration;
- Tests intended solely for use by law enforcement for forensic purposes; and
- Tests used solely for the purposes of public health surveillance.
.
LDTs Not Subject to General Enforcement Discretion
FDA also states that certain other categories of LDTs are required to comply with device requirements under the FDCA:
- Direct-to-consumer tests;
- Tests intended for use in a declared emergency;
- Tests for donors of blood or human cells, tissues, and cellular or tissue-based products; and
- Collection devices.
.
GT Takeaways from Final LDT Rule
While the phaseout policy and the new targeted enforcement discretion categories provide an extended “on-ramp” for affected LDT manufacturers, as FDA underscores in the final rule, any enforcement discretion policy is not law and can be changed by FDA at any time, without additional rulemaking, as long as the changes are “consistent with good guidance practices.” Namely, if FDA is concerned with the safety and effectiveness of any LDT already on the market, it can still require the performing laboratory to respond to inquiries and/or obtain 510(k) clearance for the LDT. FDA asserts that it will explicitly request submission of the labeling for currently marketed LDTs, to use with other information submitted under the phaseout policy, to identify LDTs that “specifically raise concerns.” This scrutiny may also be triggered by publicly available information, or a complaint, followed by an investigation conducted by FDA. Stakeholders should factor this uncertainty into their compliance planning efforts under the phaseout policy.
The final rule also leaves several items up for interpretation, especially with regard to which types of modifications would trigger the need for premarket review and how the labeling submission requirements apply to LDTs over which FDA intends to exercise targeted enforcement discretion.
Finally, while submitting an LDT for premarket review prior to the start of Stages 4 or 5 of the phaseout policy (as applicable) has the advantage of extending enforcement discretion pending a decision from FDA, the earlier submitters will need to navigate premarket review pathways (e.g., 510(k), de novo), which may involve uncertainty as to the availability of predicate devices, due to the differences between IVDs generally versus IVDs manufactured by laboratories.
Considerations for Laboratories
Laboratories must ensure that they understand all distinctions and nuances under the final rule and comply with the applicable regulations under this phaseout policy within the prescribed timeframes. The two most immediate compliance requirements for LDT manufacturers, which must be fully implemented by May 6, 2025, relate to the filing of medical device reports (21 C.F.R. Part 803), as well as notices of corrections and removals (21 C.F.R. Part 806). Prior to this first deadline, laboratories may wish to amend all policies and procedures related to the development and use of LDTs to ensure (i) proper documentation of all investigations, audits, reports, and corrective actions taken in response to any LDT-related incidents; and (ii) regular review and appropriate reporting of data related to adverse events, corrections, and removals.
Although after May 6, 2025, the next deadline is not until Feb. 2, 2026, the development and implementation of quality systems for complaint files is a multi-step process involving complex documentation and registration requirements necessitating robust organizational planning to implement. Therefore, while medical device reporting and correction and removal notices should be the first priority, laboratories should begin preparing for QMSR compliance no later than early 2025.